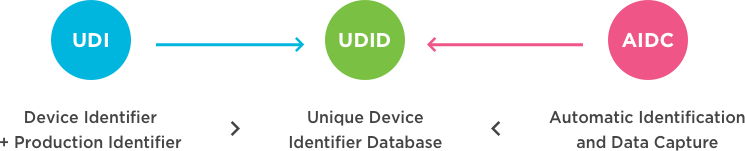
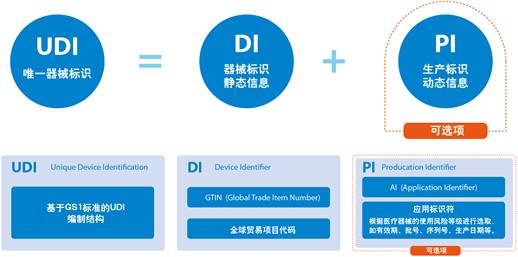
医疗器械FDA认证I,II或III类设备怎么区分?
美国FDA规范在美国销售的所有医疗器械,这些医疗器械分为三大类。FDA批准的任何医疗器械都将根据其风险,侵袭性以及对患者总体健康的影响分为I,II或III类。
但是,这三个类别之间的界限在哪里,为什么?
美国FDA的分类准则可能会使医疗器械制造商对系统的接触程度极度混乱。根据设备的分组方式,制造商的最佳市场开发途径存在巨大差异。与II类或III类设备相比,I类设备受到的法规要求要少得多。
通过了解FDA医疗器械类别中的差异,您可以了解如何对器械进行分组。掌握了这些知识后,处于上市前阶段的医疗设备制造商可以更好地准备和分配监管批准所需的资源。

FDA医疗器械类别之间的差异:
FDA已经对1,700多种不同类型的医疗设备进行了分类。这些设备按照16种专业分类在《联邦法规(CFR)》中,例如心血管或血液学设备。根据16种专业中的一种对医疗设备进行分类是了解您是否要制造I,II或III级医疗设备的第一步。
根据专业对设备进行分类后,FDA指示制造商在了解其设备是否获豁免的情况下进行上市前通知。风险和侵入性最低的I类医疗设备免于上市前通知流程。特定的II类设备也无需获得上市前批准。
但是,由FDA监管的所有设备都必须遵守有关注册,标签和质量的当前良好生产规范(cGMP)要求。但是,您如何知道您的设备是I类还是II类,以及是否需要接受售前通知?
医疗FDA认证3大分类:
1.医疗器械FDA注册1类设备:
美国食品和药物管理局将I类设备定义为“ 不旨在用于维持生命或维持生命,或在防止危害人类健康方面具有重要意义,并且可能不存在潜在的不合理疾病或伤害风险的设备 ”。
这些设备是FDA监管的最常见的设备类别,占市场上批准的设备的47%。
I类设备与患者的接触最少,对患者的整体健康影响很小。通常,I类设备不会与患者的内部器官,中枢神经系统或心血管系统接触。这些设备受最少的法规要求。
I类设备的示例:
1.电动牙刷
2.压舌板
3.氧气面罩
4.可重复使用的手术刀
5.绷带
6.医院病床
将I类医疗设备推向美国市场:
I类设备是最快,最容易推向市场的设备,因为它们给患者带来的风险最小,并且对维持生命的护理至关重要。大部分I类设备免于FDA的上市前通知(510k)和上市前批准(PMA)要求。
I类设备不受FDA通用控制的豁免,FDA通用控制是一系列适用于I,II和III类医疗设备的命令。该法案的规定涉及掺假,商标错误,设备注册,记录和良好生产规范。仍属于A类的医疗器械制造商仍必须实施质量管理体系并遵循标准以确保产品质量。
2.医疗器械FDA注册II类设备:
II类医疗设备比I类医疗设备更为复杂,并且存在较高的风险类别,因为它们更可能与患者持续接触。这可能包括与患者的心血管系统或内部器官接触的设备以及诊断工具。
FDA将II类设备定义为“ 其通用控制措施不足以合理保证设备安全性和有效性的设备”。”
II类医疗设备的示例:
1.导尿管
2.血压袖套
3.怀孕测试包
4.针筒
5.输血套件
6.隐形眼镜
7.手术手套
8.可吸收缝线
将II类医疗设备推向美国市场
控件因设备而异,但根据FDA的规定,可以包括:
1.设备性能
2.上市后监控
3.病人登记
4.特殊标签要求
5.上市前数据要求
指导方针
大多数II类设备已通过“上市前通知”或510(k)程序获得FDA批准投放市场。
II类设备受到上述相同的通用控制的约束,但FDA将其定义为“对其通用控制不足以提供合理保证该设备安全性和有效性的设备”。因此,II类设备也要接受特殊控制。这些法规取决于设备,并且可能包括特殊的标签要求,患者注册表和性能标准。
大多数II类设备通过售前通知(510k)流程进入市场。510(k)是FDA的复杂应用程序,它通过证明该设备与市场上的其他设备等效来证明该设备是安全有效的。
此过程涉及显示与另一种设备的“实质等效性”,这在FDA术语中称为“谓词”。这并不意味着设备需要相同,但是它们在使用,设计,材料,标签,标准和其他特性上需要相当的相似性。
FDA在2018年初发布了一份豁免清单,该清单免除了510(k)程序中的800多种通用I类和II类医疗设备。如果您拥有通用的II类医疗设备,则可以通过搜索FDA产品分类数据库来发现其是否免于510(k)备案。
相关阅读:彻底改革FDA 510(k)的5个理由是一个重大举措。
3、医疗器械FDA将III类设备:
FDA将III类设备定义为“通常会维持或维持生命,被植入或存在潜在的不合理疾病或受伤风险”的产品。
美国FDA监管的设备中只有10%属于III类。此分类通常扩展到永久性植入物,智能医疗设备和生命支持系统。
虽然通常将III类保留给最具创新性和最先进的医疗设备,但由于其他原因,还有其他一些设备可以归入III类。如果制造商在PMA(510k)归档过程中无法证明与谓词(现有产品)的实质等同性,则某些最初归类为II类的设备可能会升为III类。
III类医疗设备的示例:
1.乳房植入物
2.起搏器
3.除颤器
4.高频呼吸机
5.人工耳蜗
6.胎儿血液采样仪
7.植入假肢
将III类医疗设备推向美国市场:
III类设备受所有FDA通用控制和FDA上市前批准(PMA)流程的约束。FDA写道:“由于与III类设备相关的风险水平,FDA已经确定仅常规和特殊控制不足以确保III类设备的安全性和有效性。”
PMA是FDA要求的最密集的设备营销应用程序。某些FDA III类设备是免税的,并且可能符合510(k)备案的要求,但是大多数都有望获得上市前批准。
PMA流程需要对医疗器械进行严格的研究,以通过开发数据驱动的利益/风险档案来证明安全性和有效性。PMA过程通常涉及临床试验以及大量时间和资源,以进行足够的数据收集。III类中PMA流程的唯一例外是具有相当等效功能的设备。您可以通过搜索FDA 上市前批准(PMA)数据库和510(k)上市前通知数据库确定是否可以使用510(k)来销售III类设备。
怎么确定FDA注册类别?
对您的医疗设备进行分类的第一步是浏览FDA分类法规,即根据医疗专业划分的16种医疗设备类别列表。
例如,我们将向您展示识别血压报警器分类的步骤。该设备被归类为870类:心血管设备。

找到相关的医学专业后,单击类别,然后浏览设备列表,直到找到等效的内容和关联的设备代码。

单击设备代码并打开准则。设备分类在(b)节中列出。

如果您的设备在FDA分类的1700种设备中缺少列出的同等产品,则很可能是没有实质性等效产品的创新设备,将被归类为III类。
了解FDA医疗器械类别
被FDA分类为I,II或III级的医疗设备之间的差异主要在于风险,与患者及其内部系统的接触量以及设备是否对维持生命至关重要。
除了这些因素之外,FDA在确定设备的分类方式时还考虑了相当的等效性。如果您的设备风险低并且与患者的接触最少,则您很可能有资格获得I类和简化的市场批准程序。II类和III类设备必须通过实质等效性,510(k)备案或上市前批准流程证明其安全性。
通过了解您的设备如何分类,您可以通过了解FDA可能需要的过程和文件来简化获得市场认可的途径。如果您的组织受到II级或III级510(k)或PMA的要求,则此知识可以帮助您提前分配适当的资源并计划成功的申请。